


Das Spiel mit den Varianten
Widersprüche und Ungereimtheiten bei der Zulassung des BioNTech-Impfstoffes


English translation
Ich beschäftige mich seit nun über einem Jahr öffentlich auf twitter.com/a_nineties mit den von PHMPT freigeklagten Zulassungsunterlagen für BNT162b2, dem Covid-Impfstoff von BioNTech.
Im Laufe dieser Beschäftigung sind mir einige unstimmige Aussagen von BioNTech in der Öffentlichkeit und in den abgegebenen Zulassungsunterlagen aufgefallen. Dieser Artikel ist alles andere als vollständig, was Betrug, Vertuschung, und Falschaussagen seitens Pfizer/BioNTech angeht, sondern mehr eine Auflistung dessen, was mir aufgefallen ist und worüber ich mir sicher genug bin.
BNT162-01 Bekanntmachung
Beginnen wir mit diesem tweet von stefanie (@Quo_vadis_BRD):




Am 22.04.2020 gab BioNTech folgende Presseerklärung heraus. „Wir freuen uns, dass die präklinischen Studien in Deutschland erfolgreich abgeschlossen sind und wir nun bald diese erste Studie am Menschen bereits früher als erwartet beginnen werden.[..]”
Die beiden gefetteten Stellen entsprechen nicht voll den Tatsachen. Bevor ich das weiter ausführe zeigt ein Blick in das EU-Studienregister, dass die Zustimmung der Ethik-Kommission und die PEI-Genehmigung bereits am 15. bzw. 20. April vorlagen. Geschichtsträchtiges Datum.

Prä-klinische Studien
Waren am 22. April die präklinischen Studien, die in Deutschland stattfanden, beendet? Nein.
Hier ist eine Übersicht der auf PHMPT.org abrufbaren Studien:
Excel-Datei BNT Zeitleiste Stand 24.03.23
BNT Timeline English
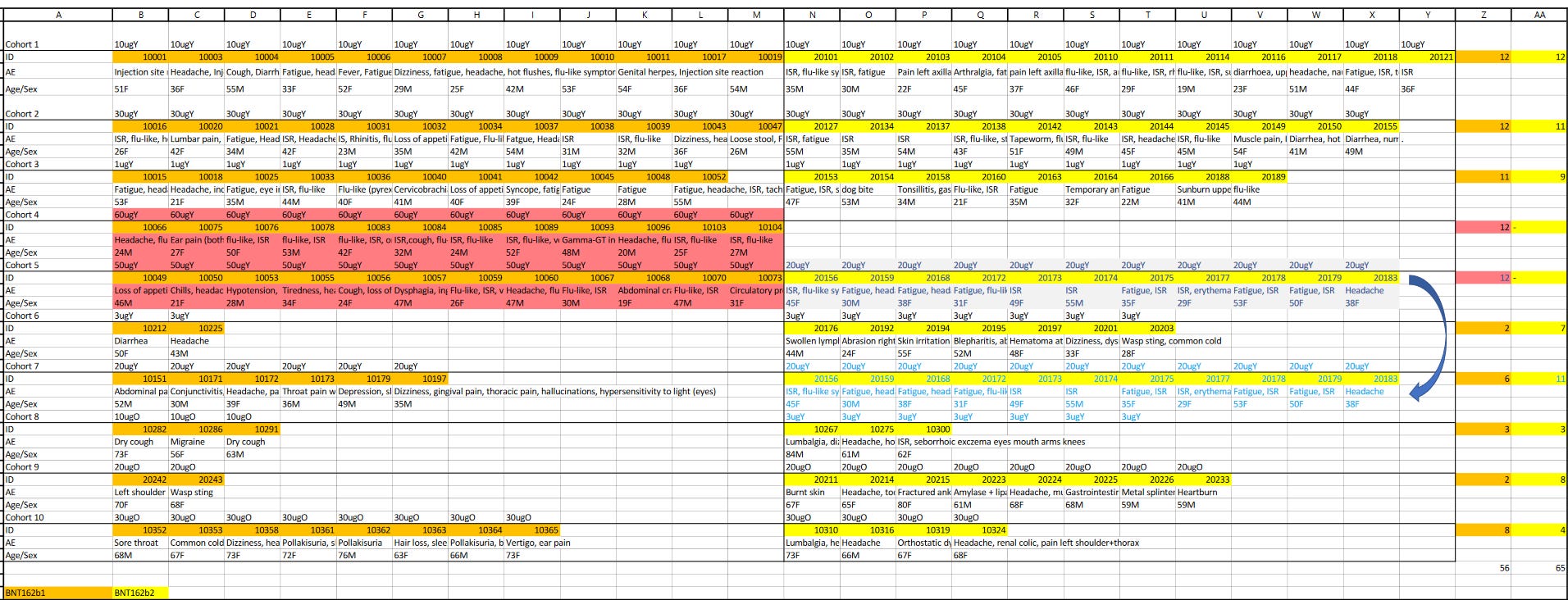
Die beiden roten Balken sind die klinischen (sprich menschlichen) Studien.
BNT162-01 (Kein placebo-Arm, Dosierungsfindung und “first in human”-Studie, 96-120 Teilnehmer je Produktversion, dann nochmal etwa 200 Patienten in drei Erweiterungskohorten für BNT162b2)
C4591001 (Placebokontrollierte und pseudogeblindete Dosierungsfindungsstudie, Phase 1/2 mit 195-840 (Endstand/zwischenzeitliche Höchstzahl) Teilnehmern, Phase 2/3 ab Ende Juli mit 44,000+)
Von 15 bislang einsehbaren prä-klinischen Studien begannen 10 nach den menschlichen Versuchsreihen.


Am 22.4.2020 war genau eine prä-klinische Studie beendet, R-20-0072. Vier weitere liefen bereits, eine davon (R-20-0085) in Deutschland, und mindestens zwei weitere fanden später im Bundesgebiet statt (R-20-0112, R-20-0211).
Die prä-klinischen Studien in Deutschland waren also nicht abgeschlossen, sondern auch in Deutschland noch in der Durchführung bzw. noch gar nicht begonnen. Wie kommt es dann zu einer Aussage, deren Inhalt kaum entgegengesetzter verstanden werden könnte?
Erste Studie am Menschen
Die Studie BNT162-01, die in der Pressemitteilung angekündigt wurde, war die sogenannte “first-in-human”-Studie. Oder sollte es zumindestens sein, denn nur der Impfstoffkandidat BNT162b1 (und vermutlich BNT162a1, zu dem es aber keine Patientendaten gibt) wurde in ihr zuerst erprobt.
Die Version, die auf den Markt gekommen ist, BNT162b2, wurde zuerst in Studie C4591001 am 8.6.2020 erprobt. c4591001-interim-mth6-randomization-sensitive.pdf
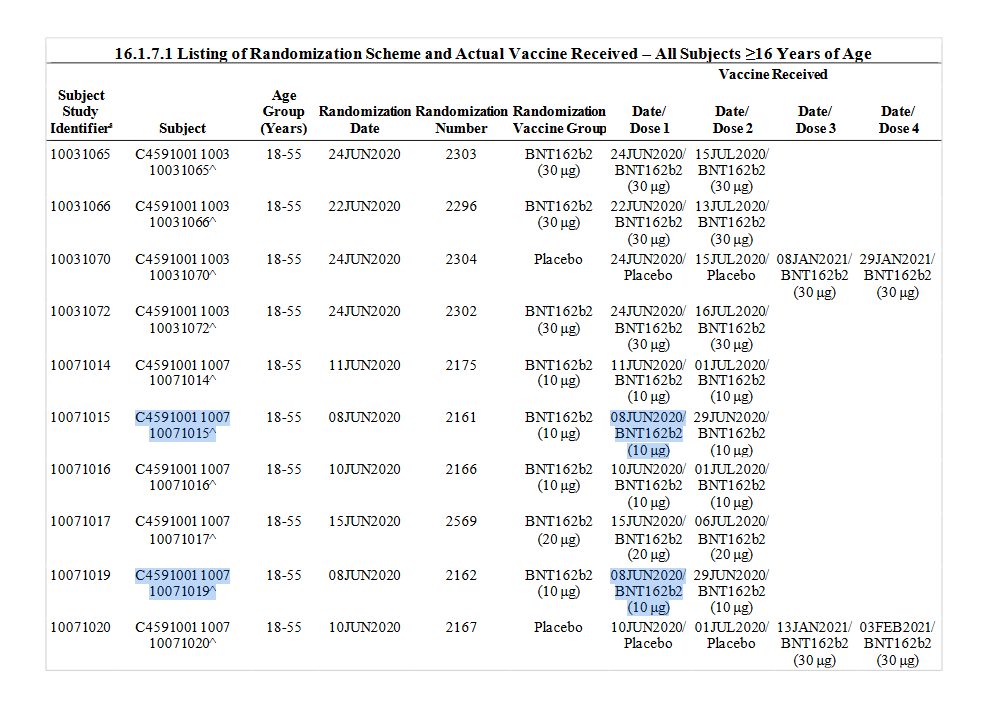
Die erste Verwendung von BNT162b2 in BNT162-01, der vermeintlichen FIH-Studie, war erst eine Woche später, am 15.6.2020. BNT162-01-interim3-patient-batches.pdf
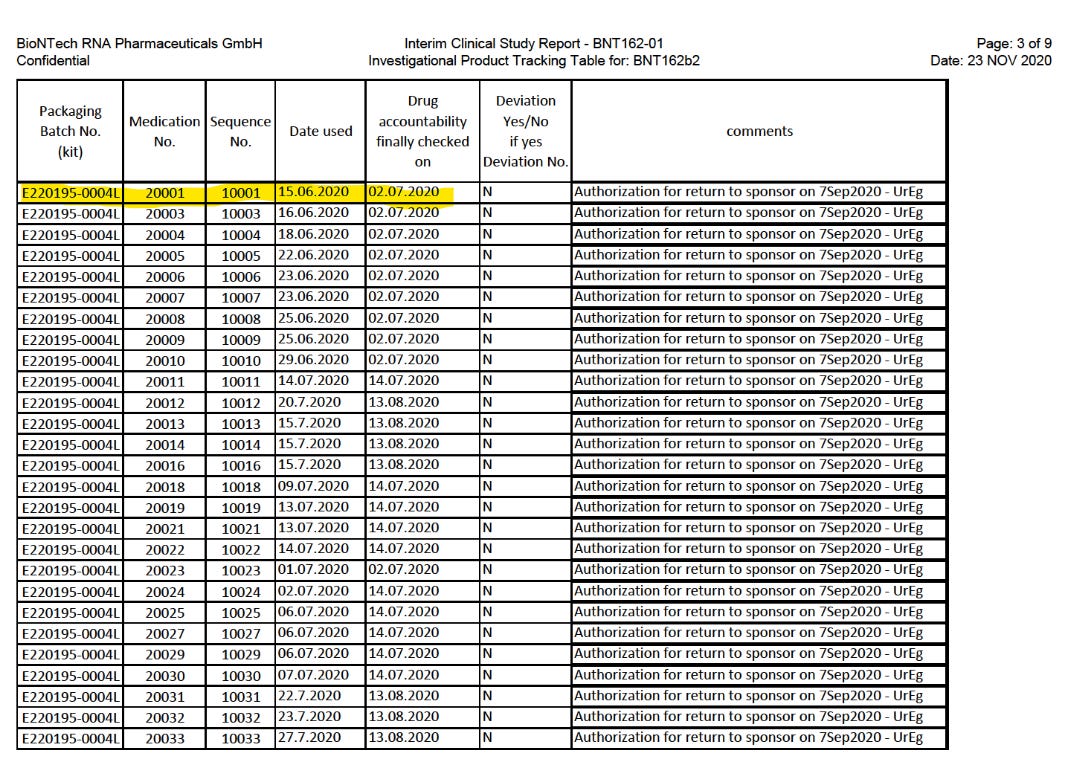
Also war BNT162-01 nur für die Kandidaten BNT162a1 (keine Daten vorliegend) und BNT162b1 die first-in-human Studie. BNT162b2 wurde nachweislich zuerst in C4591001 verwendet.
Wie ich noch zeigen werde unterscheiden sich BNT162b1 und BNT162b2 nicht nur im ausgedrückten viralen Protein, sondern auch in der Zusammensetzung, womit BNT162-01 nicht mehr die Definition der ersten Studie im Menschen erfüllen kann.
Wieso so spät?
Wieso der einzige Impfstoffkandidat, der eigene prä-klinische Studien erhalten hat, so spät im Studienverlauf erst verwendet wurde? Gute Frage. Ich lasse hier kurz ab von spezifischen Aussagen BioNTech’s und zeige Informationen aus den Unterlagen, um mich an einer Antwort zu versuchen.
Ein Paar Hinweise sind im Studienprotokoll für BNT162-01 zu finden.
Seite 12 und 17: “Die Untersuchungen an den unterschiedlichen Impfstoffen könnten aus logistischen Gründen möglicherweise nicht gleichzeitig beginnen.”


Seite 181 der PDF-Datei: Sektion 2.2.1. zeigt, dass bis 22.6, also genau sieben Tage nach der ersten BNT162b2-Gabe in BNT162-01, nur Daten zu BNT162a1 und BNT162b1 vorlagen.
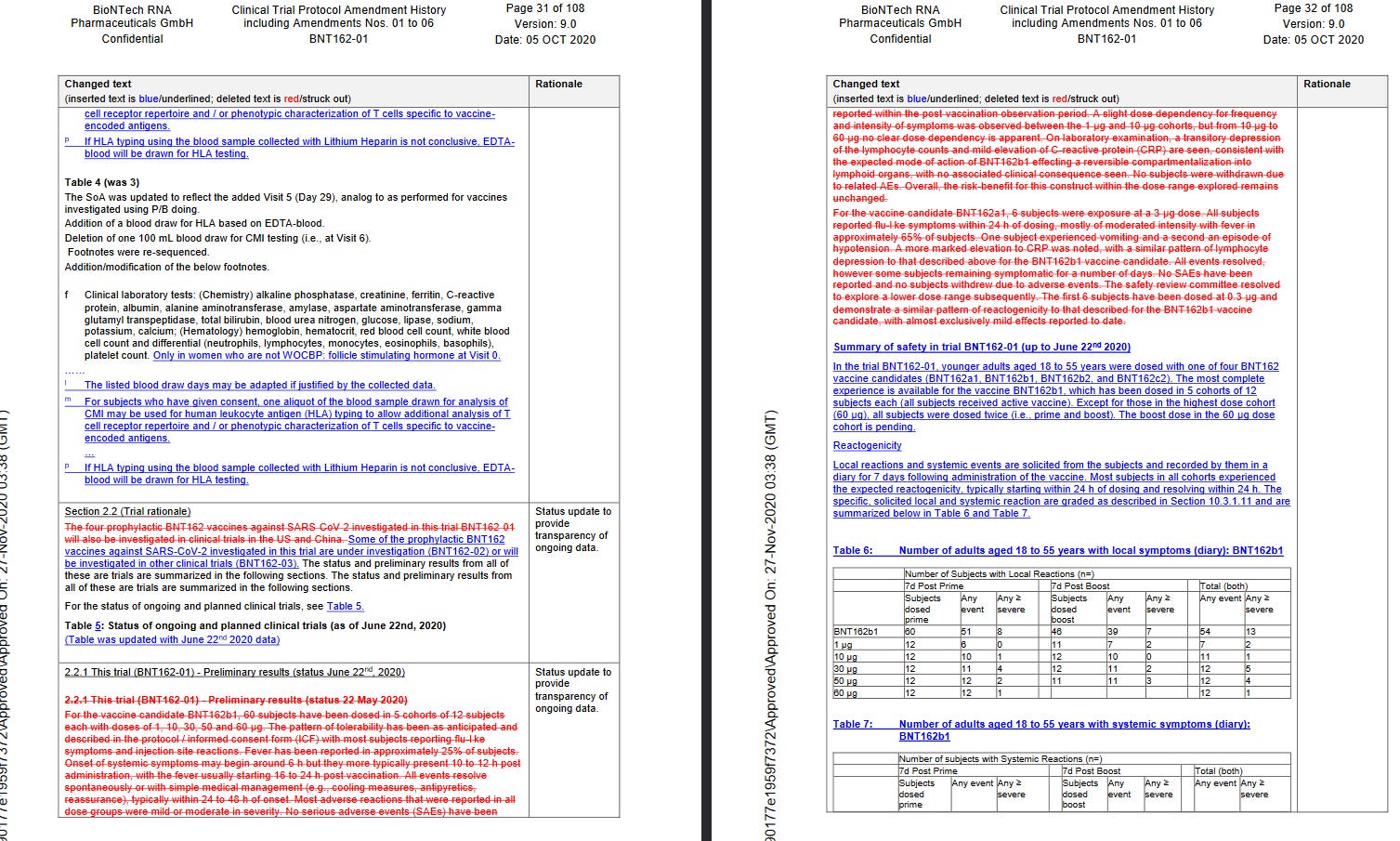
Schliesslich steht auf Seite 184 explizit, dass BNT162b2 und BNT162c2 “vor kurzem mit der Dosierung begonnen haben”. Diese Bemerkung ist besonders wichtig, weil es sonst zu c2 kaum bis gar keine Daten gibt und nur anhand der Protokolländerungen sich erörtern lässt, ob und wann es verwendet wurde.

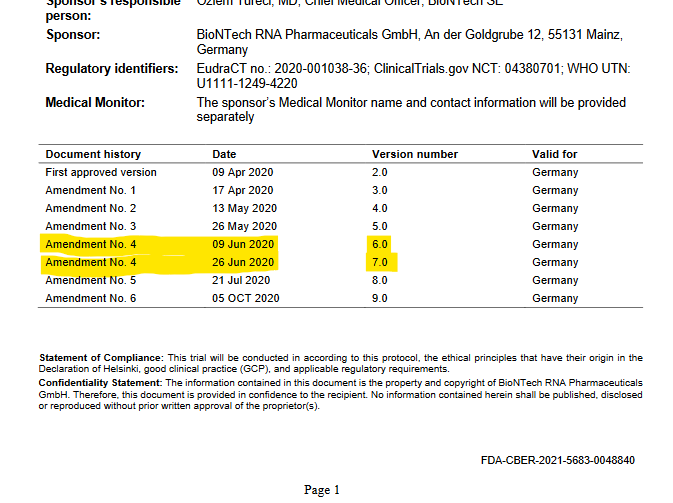
Diese Änderung des Protokolls fand zwischen dem 9.6. und 26.6.2020 statt. Wieso zwischen 9.-26.6.? Es wurden zwei Versionen des Studienprotokolls mit zwei verschiedenen Datumsangaben unter dem gleichen “Amendment” aufgeführt. Der Wort-zu-Wort Veränderungslog ab Seite 153 wird nur zwischen Amendments, nicht zwischen Versionen geführt. Damit lässt sich nicht nachvollziehen, ob eine Änderung nun am 9.6 oder am 26.6 stattfand.
Sowas schon mal gesehen? Ich nicht. Wie ist das über Monate hinweg niemandem aufgefallen, der dieses Protokoll in die Hand genommen hat?
Was für “logistische Gründe” könnten das sein, die zweimal genannt werden? Genug BNT162b2 war auf jeden Fall da, um ab Anfang April mehrere Kohorten Rhesusaffen in Pearl River, NY zu impfen (VR VTR 10671), oder ab Anfang März und Mai Mäuse in Mainz (R-20-0085, R-20-0112).
Für die 12 Personen der ersten BNT162-01 Kohorte wäre zweifelsohne genug Material dagewesen und die globale Lieferkette funktionierte offensichtlich reibungslos.
v8 und v9
Es befanden sich ausserdem zwei b2-Versionen, v8 und v9, in der Entwicklung. Studie 38166 (März 2023 auf PHMPT erschienen) ist die erste mit v8-Daten. Es gibt noch zwei weitere Studien, die sich auf v8 beziehen; diese sind noch nicht veröffentlicht, werden jedoch in diesem TGA-Dokument genannt und kurz beschrieben:
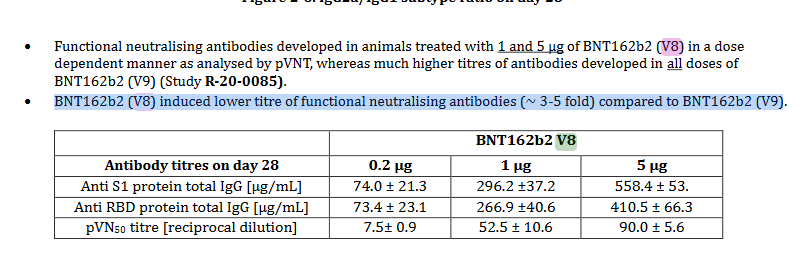
In der Zusammenfassung von R-20-0054 wird vermerkt:”BNT162b2(V8) induzierte einen (~3-5x) niedrigeren neutralisierenden Antikörperspiegel als BNT162b2(V9)”. Eine mögliche Erklärung wäre, dass v8 mit Pseudouridine (“Ψ”) und v9 mit n1-Methylpseudouridine (“m1Ψ”) kodon-optimiert wurde. Bis Studien 0054 und 0360 einsehbar sind bleibt das eine Hypothese, aber mit solidem Fundament, siehe unten ein weiteres Zitat aus o.g. TGA-Dokument, Seite 7; “BNT162b2 V8 und V9 Varianten haben identische Aminosäure-Sequenzen, mit geringen Unterschieden in der Kodon-Optimierung zwecks besserer Antigenproduktion.”.

Ausserdem gibt es meines Wissens keinen Verweis auf unterschiedlich kodon-optimierte Versionen von BNT162b1, was vermuten lässt, dass sämtliches verwendetes BNT162b1 Ψ und nicht m1Ψ verwendet [Anmerkung: b1 benutzt m1ΨTP]1, was die Vergleichbarkeit der beiden Stoffe zusätzlich in Frage stellt [stimmt trotzdem]. Dabei ist diese Annahme für die Validität der Studie als Ganzes fundamental wichtig, wie die Erstgabe von BNT162b2 in der nicht-”first-in-human”-Studie C4591001 unmissverständlich zeigt.
Die bislang einzige v8-Studie (38166) ist abgesehen von R-20-0072 (in der Luciferase-mRNA verwendet wurde, also gar kein SARS-CoV-2 Antigen) die mit dem frühesten Anfangsdatum. Alle weiteren bislang einsehbaren Studien beginnen später und benutzen v9. Es ist naheliegend, dass BioNTech innerhalb dieses Zeitraums von v8 auf v9 umgestiegen ist. Die erste Charge v9 entstand am 9. April. Dauerte es deshalb so lange, bis Menschen damit injiziert wurden?
Anmerkung: Der Unterschied zwischen BNT162b2v8 und BNT162b2v9 sind zusätzliche “cytosine ribonucleotides”.2
Oder war die Absicht eventuell ein möglichst kurzes Beobachtungszeitfenster für Nebenwirkungen zu haben? In diesem Kontext eine weitere Ungereimtheit:
Bekanntmachung Phase 3 Impfstoff
Am 27. Juli gaben Pfizer und BioNTech bekannt, dass BNT162b2 30µg in Phase 3 der Studie C4591001 untersucht werden wird. Die Protokolländerung, in der diese Entscheidung festgehalten wird, geschah schon am 24. Juli.


Dieser wesentlich spätere Beginn der b2-Dosierungen (54 Tage nach Studienbeginn in BNT162-01, 35 Tage in C4591001) und die Phase 3 Entscheidung für BNT162b2 30µg am 24.7.2020 habe ich nochmal in der Excel-Grafik veranschaulicht:
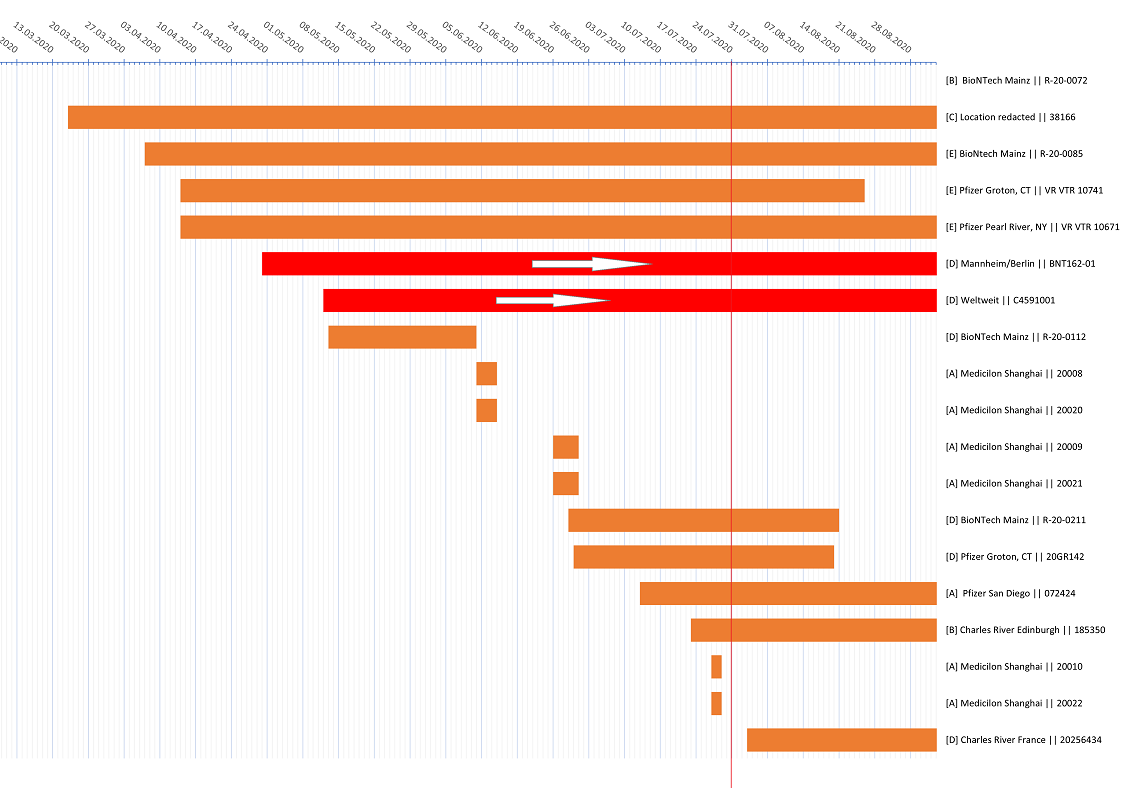
Nach Beginn der b2-Dosierungen vergingen in BNT162-01 und C4591001 jeweils 39 bzw. 46 Tage bis zur Entscheidung, das Produkt für Phase 3 zu verwenden.
Die Entscheidung wurde drei Tage vor Beginn der einzigen Entwicklungs- und Fruchtbarkeitstoxizitätsstudie getroffen.
Wieso vergehen drei Tage zwischen der Protokolländerung am 24. und der Pressemitteilung am 27.7., bevor diese wichtige, aktienkursrelevante Entscheidung öffentlich gemacht wird?
Wieso wird der Impfstoffkandidat mit der umfangreichsten zugrundeliegenden präklinischen Arbeit (z.B. der einzigen Primaten-Challenge-Studie), nach weniger als der Hälfte an beobachteter Patientenzeit verglichen zu den präklinisch geringer erforschten Varianten a1 und b1, zum Phase 3-Kandidaten gekürt? Vielleicht weil die Entscheidung von vornherein feststand?
Dafür spricht zum Beispiel, dass in Studie BNT162-01 b1 weniger Nebenwirkungen verursacht hat als b2.
Nebenwirkungen BNT162-01
Exceldatei Nebenwirkungen BNT162-01 (inkl. Rohdatensatz)
Auf den ersten Blick (80 Nebenwirkungen bei b1, 65 bei b2) scheint die Sache klar zu sein; b2 ist das sicherere Produkt. Bei näherer Betrachtung jedoch: die beiden höchsten Dosierungsgruppen für b1, 50µg und 60µg, haben keinen Äquivalent bei b2.
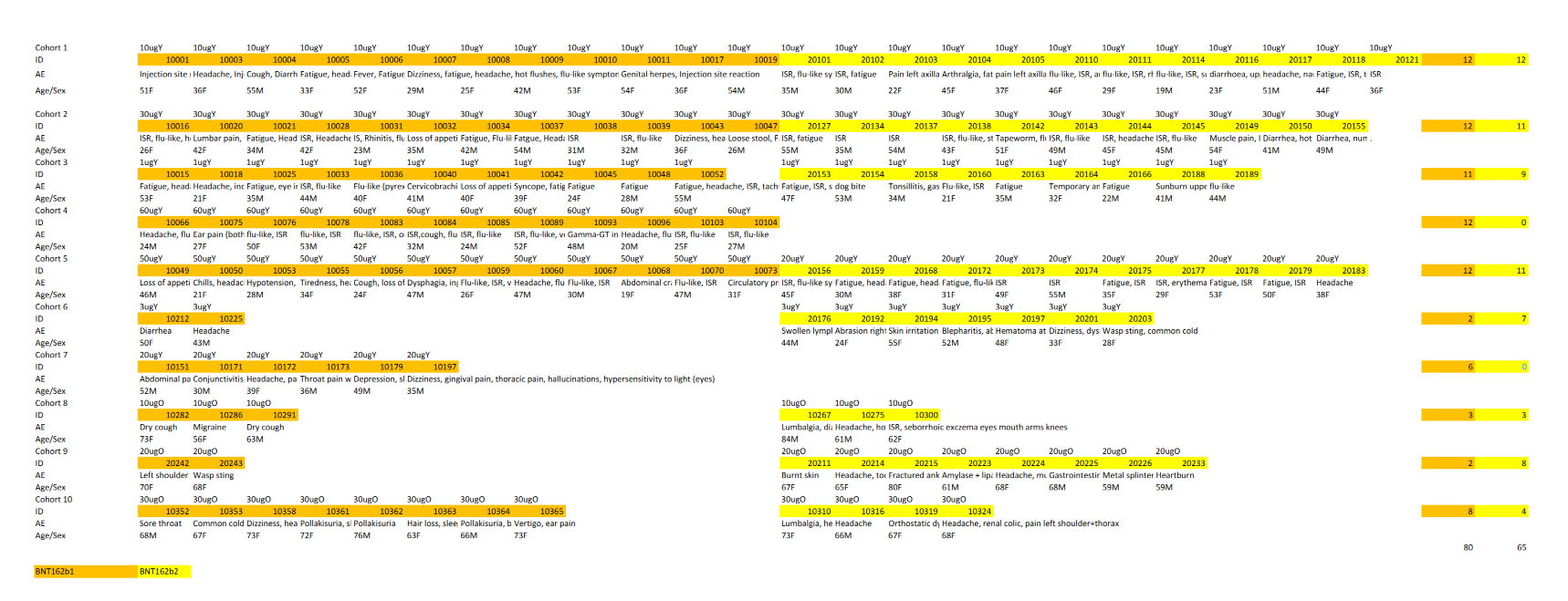
Wenn man diese 24 Patienten ausschliesst, um die Dosierungsgruppen direkt miteinander zu vergleichen, verkehrt sich der erste Eindruck ins Gegenteil. b1 hat ohne die hochdosierten Patienten “nur” 56 Geschädigte, was die 65 von b2 deutlich schlechter dastehen lässt. Beide Ziffern beziehen sich auf nur 96 Patienten je Version (8 Kohorten mit je 12 Patienten).
Es würde auch rein biologisch betrachtet wenig Sinn machen, dass das grössere, modifizierte Antigen in b2 verglichen zu dem viel kleineren in b1 sowohl weniger Nebenwirkungen verursacht, als auch mehr und stärker bindende Antikörper erzeugt. An dieser Stelle ein wichtiger Unterschied zwischen den Varianten a1/b1/a2/c1 und b2/c2, nämlich RBD (receptor-binding domain) vs. volles Spike mit zwei Prolin-Mutationen (2P-S, Sp2, S-2P).
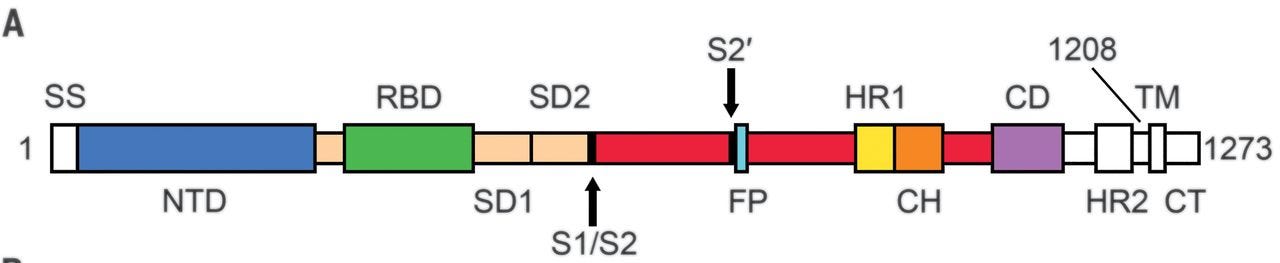
BNT162a1/a2/b1/c1 kodieren lediglich den grünen Teil der oberen Abbildung. BNT162b2/c2 kodieren das Spike in voller Länge, also das gesamte abgebildete Protein und die ganzen Probleme, die damit einhergehen.
Um das bislang Erläuterte damit in Kontext zu setzen: die 2P-S-Produktvarianten waren das ganz offensichtlich ambitioniertere und riskantere Unterfangen verglichen zu den RBD-Entwürfen. Wieso wurde die klinische Phase 1/2 durch die extrem verspätete menschliche Verabreichung so kurz gehalten, und was genau sind die “logistischen Gründe”, die für die asynchrone Untersuchung der Kandidaten unzureichend und anmassend vorgehalten wurden?
Das Paul-Ehrlich-Institut und das volle Spike
Im Studienprotokoll für BNT162-01 verrät die erste Protokolländerung vom 17. April:
”Diese Protokolländerung beschreibt das Ersetzen des Produktcodes BNT162c1 durch BNT162c2. Die RNA-Komponente in BNT162c1 kodierte lediglich die RBD des Spikeproteins, die RNA-Komponente in BNT162c2 kodiert eine modifizierte Version des Spikeproteins in voller Länge. BNT162c1 wurde durch BNT162c2 wegen dessen überlegenem Immunogenitätsprofil in nichtklinischen Studien ersetzt.
Diese Protokolländerung beschreibt Änderungen, die nach Feedback des deutschen PEI gemacht wurden (16. April, 2020). […]"

Von welchen nicht-klinischen Studien ist hier die Rede? Bis 17. April hatten folgende nichtklinischen Studien begonnen:
R-20-0072, 14.1. - 23.1.2020
Material: BNT162-LNP-Mischung mit Luciferase-mRNA beladen, um in den Versuchstieren Biolumineszenz zu messen.38166, 16.3. - 17.9.2020
Material: BNT162a1, a2 (aus Versehen, anscheinend), b1, b2v8 und c1.R-20-0085, 31.3. - 17.9.2020
Material: BNT162b2v9.VR VTR 10741, 7.4. - 19.8.2020
Material: BNT162b2v9 RNA aus DNA-Plasmiden (ohne LNP).VR VTR 10671, 7.4. - 1.11.2020
Material: BNT162b2v9.
Darüber hinaus gibt es mindestens drei weitere, noch nicht öffentliche Studien, auf die das TGA-Dokument verweist: R-20-0054, R-20-0360, R-20-0357. Keine dieser Studien hatte BNT162c1 oder BNT162c2 als Gegenstand.
Die erste prä-klinische Studie mit BNT162c2 ist R-20-0112, 6.5. - 4.6.2020.
Wenn es bis 17. April keine prä-klinische Studie mit BNT162c2 gegeben hat, kann auch kein “überlegenes Immunogenitätsprofil” aus nicht-erhobenen Daten von nicht-behandelten Tieren entstehen. Weswegen also der Wechsel von c1, RBD, zu c2, vollem Spikeprotein?
Und welche Rolle hat das PEI dabei gespielt? Auf deren Webserver befindet sich eine “Presse-Briefing 22.04.2020” betitelte PDF-Datei von “Klaus Cichutek et al.”, die zehn Präsentationsfolien zeigt, unter anderem die untere.
Schon fast putzig, wie präzise die Pfeilchen eingezeichnet wurden; dabei stellen sie die direkte und wiederholte Einflussnahme der zuständigen Zulassungsbehörde auf die Entwicklung eines Produkts dar, dass laut Herstellerangaben vom 31.3.2020 sowohl von der amerikanischen FDA3 als auch der europäischen EMA4 als gentherapeutisches Medizinprodukt eingeschätzt wurde.
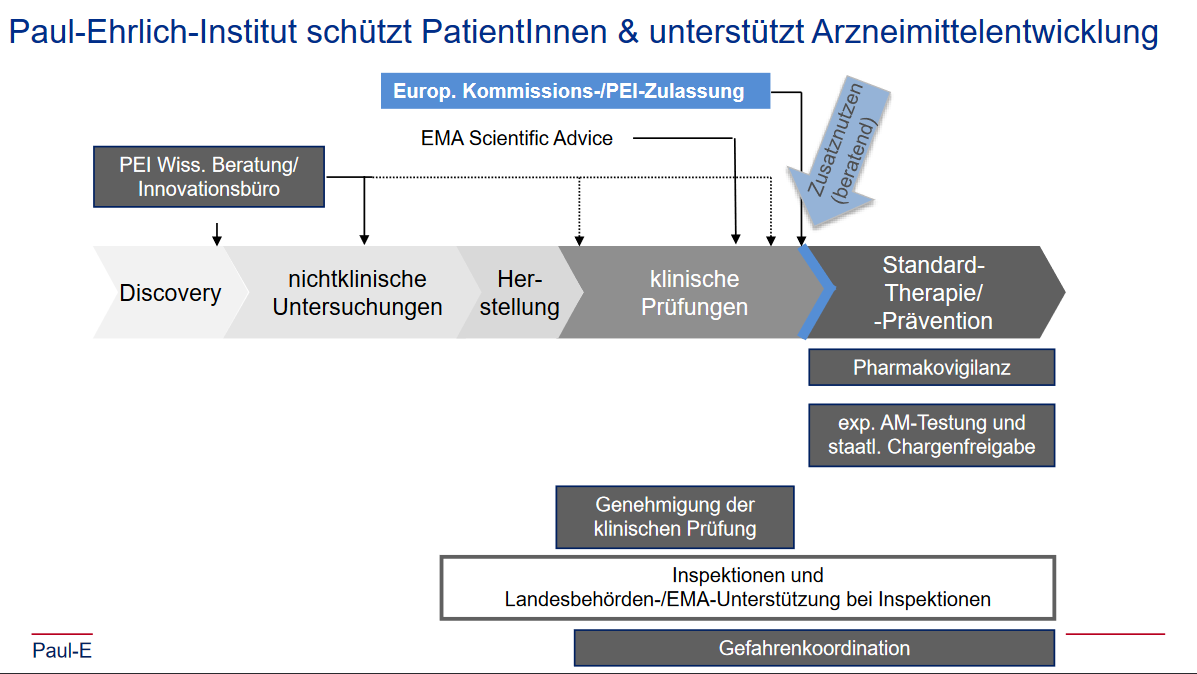
Ich habe für diese wissenschaftlichen Beratungssitzungen seit Monaten Informationsfreiheitsgesetzanfragen laufen, einzusehen hier und hier. Das Mängelschreiben vom 16.4.2020 hab ich zwischenzeitlich in teils geschwärzter Form erhalten.
Diese Beratungstreffen werden auch im Protokoll angesprochen (die Kommunikation 16. April hatte ich bereits erwähnt), BNT162-01: “Das ausgewählte Studiendesign spiegelt Diskussion und Ratschläge seitens des PEI wieder, die in wissenschaftlichen Beratungssitzungen Februar, März, und Juni 2020 im Angesicht einer sich rapide verändernden Situation entstanden.” Irgendwie versäumt BioNTech an dieser Stelle zu erwähnen, dass im April zwei weitere Feedbacks vom PEI kamen.
Ebenfalls auf dem PEI-Webserver befindet sich die BioNTech PDF “Update on Clinical Program April 2020”. Neben Falschangaben zu Beginn der Forschungsarbeiten (R-20-0072 begann am 14.1.2020) und Zustimmung der Ethik-Kommission & PEI (15.4. und 20.4.2020, weder noch am 21.4.) sind die Daten der ersten drei Treffen mit dem PEI genannt; 6.2.2020, 20.3.2020, und 8.4.2020.

Also gibt es Hinweise auf insgesamt fünf Treffen zwischen PEI und BioNTech:
6. Februar
20. März
8. April
16. April
Tag unbekannt Juni
Wenn man sich in die Lage von BioNTech versetzt, welchen Wert würde man “Ratschlägen und Diskussionen” beimessen, die dem zuständigen nationalen Zulassungsgremium entspringen? Die Behörde, dessen Zustimmung die grundlegende Voraussetzung für jegliche Zukunft des Produkts, sogar der Firma ist? An dieser Stelle noch ein Zitat von Klaus Cichutek (Danke nochmal an @quo_vadis_BRD) , jahrzehntelanger Chef des Paul-Ehrlich-Instituts:
“Wir sind die EMA”
Chargen aus der Zukunft
Wer genau aufgepasst hat, hat die Unstimmigkeit bemerkt: die Experimente beginnen oft, bevor die im Dokument angegebene Charge freigegeben ist. Studie 38166 ist in bizarrem Ausmaß davon befallen: von 10 Behandlungsgruppen werden 8 vermeintlich vor dem 26.3.2020 eindosiert.


Bloss sind die Certificates of Analysis der verwendeten Materialen handschriftlich auf 26.3.2020 datiert.
Polymun Scientific in Klosterneuburg, Österreich hat für die klinischen Versuchsreihen die RNA von BioNTech in LNPs geladen, und befindet sich jeweils ~780km und ~880km entfernt von den BioNTech-Standorten in Mainz und Idar Oberstein. Ich sehe nicht, wie man argumentieren könnte, die Dokumente seien woanders unterschrieben worden, als auf dem Briefkopf geschrieben steht, und dass sie das Ende des Herstellungsprozesses und die Freigabe seitens Polymun formalisieren.
Womit wurden dann die Tiere in 38166 am 17., 23., und 24.3. injiziert, wenn am 26.3. die Chargen erst von Polymun in Österreich freigegeben wurden?
Die Entblindung der Phase 3
Die Marktzulassungen (ob CMA, EUA etc. unterscheide ich hier nicht) BNT162b2 von den jeweils zuständigen Behörden waren:
Grossbritannien MHRSA, 2.12.
USA FDA, 10.12
EU EMA, 21.12
Dieser wichtige Artikel im British Medical Journal zeigt, dass bereits am 17.12. Pfizer Briefe an Studienteilnehmer verschickte, in denen darauf hingewiesen wurde, dass die Blindung auf Wunsch aufgehoben werden kann. Ausserdem gab die Behörde bekannt, dass die Entscheidung zur Entblindung den Herstellern überlassen wird.
Nach einem Hinweis von stefanie habe ich herausgefunden, dass Pfizer den Studienteilnehmern bereits am 10.11, einen Tag nach Veröffentlichung der ersten Interimsanalyse, einen Brief geschickt hat, in dem beschrieben wird, dass die Firma bereits an Wegen arbeite, wie die Blindung aufzuheben sei.5
Diese Entscheidung wurde bereits am 1.12.2020 getroffen, also vor jeder Marktzulassung. Protokolländerung 10:

Die erste BNT162b2-Gabe an einen Placebo-Patienten war am 14.12.2020, Patient 11521564.
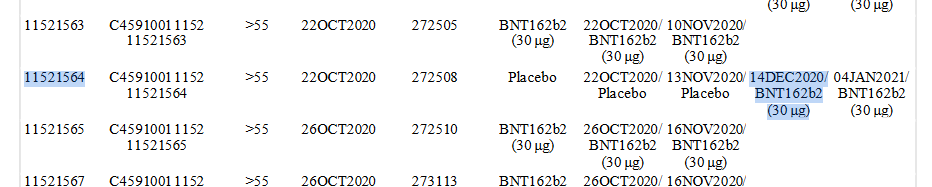
Bis 13.3.2021 (Daten-Endpunkt) hatten 85% der Placebo-Patienten eine Impfung erhalten, nachdem sie im Durchschnitt 97 Tage in der Studie verbracht haben.
Zusammenfassung
Die 2P-S Impfstoffe BNT162b2 (v9) und BNT162c2 wurden präklinisch am meisten erforscht, dann jedoch in der klinischen (menschlichen) Phase 1/2 nach immenser und unbegründeter Verspätung verwendet, am kürzesten beobachtet, und Ersterer aus intransparenten Gründen zum Kandidat für Phase 3 ausgewählt.
Die deutsche Regierung hat direkten, wiederholten und teilweise maßgeblichen Einfluss auf alle Stufen des Entwicklungs- und Zulassungsprozess genommen.
In den Zulassungsstudien finden sich viele und vielfältige Widersprüche, unstimmige Datensätze, und zeitlich unmögliche Zusammenhänge.
Die Zulassungsunterlagen haben noch nie irgendeiner Form der Überprüfung standhalten können. Das universelle, koordinierte Durchwinken dieser Produkte anhand dieser Beweislage ist allein im Anbetracht der ungeheueren Anzahl verabreichter Einheiten das größte, systematischste Verbrechen der Menschheitsgeschichte.

https://www.nature.com/articles/s41586-020-2814-7#change-history
BNT162b1 benutzt wohl doch n1-methylpseudouridin. Die Annahme besteht aber weiterhin für BNT162b2v8 und v9.

V8 and V9 have identical amino acid sequences of the encoded antigens and differ only in their codon optimisation sequences, which were designed to improve antigen expression (V9) with higher content of cytosine ribonucleotides. https://www.tga.gov.au/sites/default/files/foi-2389-06.pdf#page=18&zoom=auto,-278,174
Zitat BioNTech SEC-Formular 20-F, freigegeben 31.3.2020, Seite 16

Zitat BioNTech SEC-Formular 20-F, freigegeben 31.3.2020, Seite 26

Zum Verhältnis von Ugur und Klaus Cichutek habe ich schon was aus Lightspeed analysiert. Das ist einer meiner Schwerpunkte für einen abschließenden Essay. Cichutek hängt wohl auch mit Patenten in der Sache drinnen, da habe ich mich aber noch nicht durchgearbeitet, bzw. wäre nett, wenn sich jemand mit Ahnung in dem Bereich mal Klaus Patente vornehmen könnte.
https://patents.justia.com/inventor/klaus-cichutek
In Kapitel 2 findet man viel zum privaten und längeren freundschaftlichen Verhältnis zwischen Klaus und Ugur https://drbine.substack.com/p/project-lightspeed-ironieeinmal-mit-83e
„Ugur hatte ein kollegiales Verhältnis zu dessen Präsidenten, dem Biochemiker Klaus Cichutek. [… ]Am Dienstag […] griff Ugur zum Telefon und rief Cichutek direkt an.“ (S. 71)
Männerfreundschaften sind doch etwas Tolles. „In seinem Telefongespräch mit Cichutek unterstrich Ugur, dass er den Ausbruch des Coronavirus extrem ernst nehme,[…]“ (S. 71).
„Ein früher Pionier experimenteller Therapien, versprach Cichutek, alles zu tun, um zu helfen“ (S. 72) und Ugur wusste „Bei „gutem Willen“ der Genehmigungsbehörden, hatte Ugur zu Kollegen gesagt, könnten schon Ende des Jahres weltweit Spritzen […] Menschen zugutekommen.“ (S 72).
Cichutek war auch mit der Aufkärung des Jesse Gelsinger Falls 1999 beuaftragt...
Das erklärt vielleicht ein wenig vom Verhalten des PEI.
Die haben übrigens auch ein gemeinsames Paper, wie man mRNA-Plörre in der EU zulässt auf Sonderwegen:
Hinz T, Kallen K, Britten CM, Flamion B, Granzer U, Hoos A, Huber C, Khleif S, Kreiter S, Rammensee HG, Sahin U, Singh-Jasuja H, Türeci Ö, Kalinke U. The European Regulatory Environment of RNA-Based Vaccines. Methods Mol Biol. 2017;1499:203-222. doi: 10.1007/978-1-4939-6481-9_13. PMID: 27987152.
!Wieso wird der Impfstoffkandidat mit der umfangreichsten zugrundeliegenden präklinischen Arbeit (z.B. der einzigen Primaten-Challenge-Studie), nach weniger als der Hälfte an beobachteter Patientenzeit verglichen zu den präklinisch geringer erforschten Varianten a1 und b1, zum Phase 3-Kandidaten gekürt? Vielleicht weil die Entscheidung von vornherein feststand?!
Lösung aus Project Lightspeed (das Kapitel, an dem ich gerade interpretiere)
„Nach jener Videokonferenz, in der Uğur die meisten Entscheidungsbefugnisse abgegeben hatte – was Roshni Bhakta, die Leiterin des Bereichs strategische Partnerschaften und Lizenzen bei BioNTech, damals dazu veranlasst hatte, still in sich hineinzufluchen –, hatte er sich als eines der wenigen Rechte das letzte Wort bei der Auswahl der Kandidaten für die Phase-III-Studie vorbehalten.“ (S. 271)
Es geht eine Weile hin und her beim Zoom mit Pfizer am 24 Juli 2020 und Dormitzer, der schon vorher mit seinen Analysen recht hatte mahnt erneut zur Vorsicht „‹Seht mal, was wir da vor uns haben, kennen wir weniger gut, aber es hat mehr Daten, die in die richtige Richtung weisen .›»“ Dem Typen kann man wohl nichts vorwerfen.
„Von den zwanzig Kandidaten, die im Februar das Labyrinth des «Projekts Lightspeed» betreten hatten, waren vier klinisch getestet worden. […] Würde das Virus einen Mechanismus entwickeln, um sich dem Zugriff des Immunsystems zu entziehen? B2.9, sagte Ugur damals zu seinen Kollegen, sei «so gut, wie es kaum besser geht», um eine spezialisierte Streitmacht zu mobilisieren: Antikörper und T-Zellen. Aber es bestand immer noch eine Chance, dass SARS-CoV-2 das evolutionäre Wettrüsten gewinnen würde. «Wir kennen die andere Seite der Gleichung nicht», sagte Ugur. «Wir wissen nicht, wie sich der Feind verhalten wird.»“ (S. 272)
Ugur hat's laut "Project Lightspeed" so im Alleingang entschieden, weil er es halt besser weiß als Leute mit Erfahrung.