


Betrug unter Aufsicht
Ein Blick durch die Lupe auf die Teleskopierung

English translation available here:

Dieser Artikel soll dazu dienen, meine Erkenntnisse auf Deutsch zusammenzuführen, und ausserdem die Aussagen in meinen Corona-Ausschuss-Interviews1 zu untermauern. Die PHMPT FOIA-Produktion der BNT162b2-FDA-BLA-Lizenz für Ü16 soll Dezember 2023 beendet sein2; anders formuliert befinden sich in den zwei noch ausstehenden Produktionen die ~150.000 brisantesten Seiten - ein guter Zeitpunkt um Revue passieren zu lassen. Leider gibt es in einem Artikel nur eine begrenzte Menge Platz, ich habe viel auslassen müssen.
[19.11.2023]: Den Abschnitt um Prozess 2 erweitert.

Die BioNTech-Acuitas-Vorarbeit in 2019

Unter dem verfügbaren Studienmaterial ist bislang Studie R-20-00723 das früheste Dokument. Es beschreibt die Bioverteilung mehrerer Luziferase-modRNA-LNP- Konstrukte in BALB/c-Mäusen (Albino-Laborrasse). Luziferase ist ein bioluminiszentes Peptid, das u.a. in Glühwürmchen vorkommt; dessen Expression kann mit Kontrastmittel und besonderem Bildgebungsverfahren in vivo sichtbar gemacht werden. BioNTech hat die “drug substance” oder DS, also modRNA, die Luziferase-Protein ausdrückt, zur Formulierung zum “drug product” oder DP an Acuitas geschickt.
Laut den “Certificates of Analysis”4 oder Analysezertifikaten entstand die BioNTech Luziferase-modRNA zwischen 27.05.2019 und 11.06.2019. Ersteres ist das Bestelldatum, letzteres findet in der Chargenbezeichnung Erwähnung (RNA-EH190611-01c). Die Agilent-Qualitätskontrolle ist auf 14.06.2019 datiert.
In den Anhängen befinden sich auch die Acuitas-Lieferscheine5. Die letzte Tabelle ist unzureichend geschwärzt, und mit Bildbearbeitungssoftware kann der Inhalt leserlich gemacht werden6. Daraus geht hervor, dass ausser RNA-EH190611-01c eine weitere, frühere modRNA-Charge von Acuitas formuliert wurde, mit der Bezeichnung “FK190222-01c”. Die strukturelle Ähnlichkeit in den Bezeichnungen lässt die Schlussfolgerung zu, dass die Charge am 22.02.2019 entstand.
Der erste Acuitas-Lieferschein ist auf 26.11.2019 datiert. Diese Datumsangabe lässt vermuten, dass die Lieferung für die “ApoE-Knockout”-Mausstudie7 gedacht war. Dann ging alles sehr schnell, bzw. stellte sich ALC-0315 als klarer Favorit heraus, denn der zweite Acuitas-Lieferschein mit den R-20-0072-Formulierungen ist auf 11.12.2019 datiert und Herstellungsdatum 9.12.2019. Lediglich 14 Tage zwischen den ApoE und R-20-0072 Lieferungen während die Luziferase-modRNA seit Juni verfügbar war verleitet zur Vermutung, dass ALC-0315 von vorneherein entschieden war und die von Acuitas durchgeführte “Pilotformulierungsstudie” mit ALC-0218 lediglich ein Feigenblatt, um den Schein einer stattfindenden Entwicklung zu wahren.
In diesem Kontext wichtig zu erwähnen ist die Juli 2018 eingegangene Kooperation8 zwischen Pfizer und BioNTech zwecks Entwicklung eines Grippe-Impfstoffes. BioNTech’s Börsengang war erst September 2019, und dementsprechend bezüglich Cashflow in relativ bedenklicher Lage als die Lizensierung der Acuitas-Technologie stattgefunden haben muss; anhand der oben beschriebenen Februar-Charge erkennbar, fand diese Lizensierung in sehr kurzem Abstand zur Vereinbarung mit Pfizer statt.
Die Comirnaty-Abmachung mit Pfizer Datum 17.03.2020 ist wohlbekannt; weniger geläufig ist die Vereinbarung mit Shanghai Fosun zur Entwicklung und Vertrieb des Produkts in Süd-Ost-Asien am 16.03.20209. Die enge Konstellation lässt Verhandlungen im Hintergrund vermuten, und die Fosun-Unterschrift könnte durchaus als Druckmittel auf Pfizer gesehen werden. Dazu kommt die deutlich geringere Profitmarge bei Pfizer, als bei BioNTech. Die Vermutung liegt nahe, dass Pfizer den sprichwörtlichen Braten gerochen hat und sich mit der 2018-Vereinbarung einen Stück des Kuchens sichern wollte; BioNTech hat jedoch einen Haken geschlagen und hinter Pfizer’s Rücken auf eigene Faust mit Acuitas die Entwicklung einer modRNA-LNP-Plattform vorangetrieben, und Pfizer hatte März 2020 keine andere Wahl, als sich den Gegebenheiten anzupassen und mit BioNTech eine neue Vereinbarung unter weniger-als-idealen Bedingungen10 einzugehen.

BNT162b2 v8/v9 - Absichtlich falsch gemacht?

Von BNT162b2 befanden sich folgende zwei Versionen in der nicht-klinischen Entwicklung:
BNT162b2 v8: modRNA, die das prolin-mutierte, volle SARS-CoV-2 Spikeprotein kodiert, mit hauseigener BioNTech-Kodonoptimierung.
BNT162b2 v9: modRNA, die das prolin-mutierte, volle SARS-CoV-2 Spikeprotein kodiert, mit der “konsensus-optimalen”-Kodonoptimierung.11
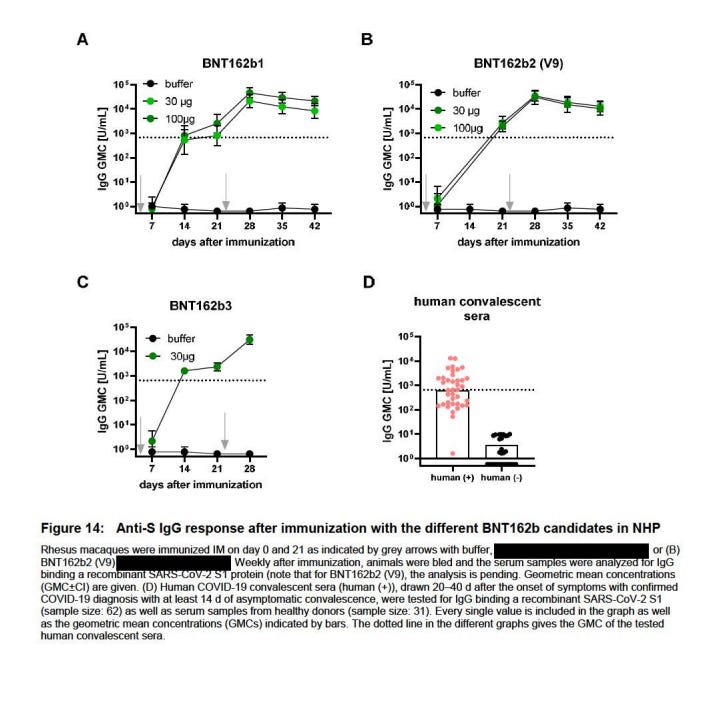
Unterlagen gibt es bislang nur zu einer BNT162b2 v8 Studie, nämlich 3816612, deren Vorabdaten für die Dosierung im Menschen erforderlich waren. BNT162b2 v9 erzeugt laut TGA 3x-5x mehr neutralisierende Antikörper13 als v8, und besitzt “zusätzliche Cytosin-Ribonukleotide”14. Am Menschen wurde nur BNT162b2v9 verwendet.
Die Aussagen in dem TGA “Nonclinical evaluation report” basieren auf zwei weiteren nichtklinischen Studien, die von der FDA noch nicht preisgegeben wurden, R-20-0054 und R-20-0360.
Die nichtklinischen Chargen hatten jeweils zwei Fertigstellungstage, einmal für drug substance (DS), die BioNTech-modRNA, und für drug product (DP), die fertige modRNA-LNP-Formulierung. Alle nichtklinischen Chargen wurden von Polymun in Österreich formuliert.
Es wurde nur eine Charge b2 v8 hergestellt, und zwar am 11.03.202015. Die nächste gefertigte Charge war die erste Charge b2 v9, Fertigstellungsdatum 19.03.2020.16 Allein daran ist zu erkennen, das b2 v8 nie ein tatsächlicher Kandidat gewesen sein kann. Es war nicht genug Zeit, um b2 v8 zu erproben und darauf zu kommen, dass man vielleicht doch besser die optimal kodon-optimierte Sequenz v9 nehmen sollte; laut dem Analysezertifikat der Studie 38166 stand das LNP-formulierte b2 v8 erst am 26.03.2020 zur Verfügung, eine Woche nachdem die erste v9 Charge hergestellt wurde.
Das bringt uns zum nächsten Widerspruch; laut Studie 38166 wurden die Versuchstiere am 17. und 24.3.2020 bereits dosiert17. Die Analysezertifikate behaupten jedoch, die entsprechenden Chargen wurden erst am 26.3. von Polymun freigegeben18. Die Distanz zwischen Hamburg, wo die Studie durchgeführt wurde, und Polymun in Klosterneuburg, Österreich macht den Widerspruch nicht geringer.
Wieso überhaupt b2 v8 für die 38166 Studie verwenden, wenn zur gleichen Zeit der eigentliche Kandidat zur Verfügung gestanden hätte? Hier gibt es eine weitere Diskrepanz zu berücksichtigen: b2 v8 hatte eine DS→DP-Zeit von 15 Tagen. b2 v9 hat dafür 21 Tage gebraucht(19.3.2020-9.4.2020), obwohl es nicht wie v8 mit drei weiteren Varianten gleichzeitig fertiggestellt wurde. Wurde Polymun gebeten, die LNP-Formulierung von b2 v9 um ein paar Tage hinauszuzögern, um die Verwendung von b2 v8 in 38166 zu rechtfertigen? Weniger abstrus als ein Experiment, dass nicht fertiggestellte Chargen injiziert.
Unmöglich ist das angegebene Zeitschema ebenfalls in Studie R-20-0085. Wie können die Experimente 9 Tage19 vor Fertigstellung der verwendeten Charge20 beginnen? Beim Verfassen dieser Arbeit bin ich darauf gestossen21, dass auch die Rhesusaffen-Studie VR-VTR-10671 mit Chargen aus der Zukunft gearbeitet hat22 - und zudem immense Unterschiede zum entsprechenden Nature-Artikel aufweist: die PHMPT-Version hat keine BNT162b1-Versuchstiere, aber dafür eine zweite Dosierungskohorte mit b2 30ug, die nicht in der Nature-Veröffentlichung auftaucht, und nur drei statt neun Tiere in der Kontrollgruppe. Verblüffenderweise enthält die “Investigator’s Brochure” (Leitfaden für Studienpersonal) Rhesusaffen-Antikörper-Messungen für drei BNT162-Impfstoffkandidaten23. Pfizer-BioNTech haben b1+b2+b3 erprobt, bei Nature die Daten für b1+b2 eingereicht, bei den Zulassungsbehörden nur b2, und es drauf ankommen lassen, dass die Studienbroschüre die zurückgehaltenen Daten verrät.
Ergebnis war, dass die Tox-Studie über b2 v8 die Humandosierung von b2 v9 rechtfertigte. Die DART-Studie zu b2 v924 dosierte erst am 27.07.2020, also am gleichen Tag, an dem Phase 3 eingeleitet wurde und ~44,000 Menschen b2 v9 30ug in Verhältnis 1:1 mit Placebo verabreicht bekamen. Etwas an die Wand fahren ist auch Teleskopierung. Die erste Verabreichung von b2 am Menschen war am 8.6.2020 in Studie C4591001, nicht in der vermeintlichen “first-in-human”-Studie BNT162-01, in dieser wurde die erste b2-Dosis am 15.6.2020 verabreicht.

Alles für den Spike

Die Entscheidung, das volle Spike-Protein zu verwenden, ist mit zunehmender Akteneinsicht intransparenter. Seit der Oktober-Veröffentlichung auf PHMPT ist die “meeting-correspondence”-Datei25 verfügbar. Unter anderem geht daraus hervor, dass Donna Boyce, ihres Zeichens “Senior Vice President Global Regulatory Affairs at Pfizer” bereits am 26.06.2020 nach einem “Typ C”-Treffen (vermutlich Chefetage) CBER26 gegenüber klarstellte, dass BNT162b2 (v9, bloß der Vollständigkeit halber, v8 war nur im nichtklinischen Bereich) der Phase 3 Kandidat sein sollte. Diese Aussage wurde am 01.07.2020 schriftlich eingereicht, und am 08.07.2020 stand die Dosis fest.

Meine bisherige Annahme, dass die Entscheidung zu b2 statt b1 auf Daten basierte, war falsch. Achtzehn Tage nach der ersten Anwendung am Menschen (8.6.2020-26.6.2020) stand der Phase-3-Kandidat effektiv schon fest, und einen Monat nach der ersten Anwendung im Menschen verpflichtend bei der FDA eingereicht.
Im Vergleich hatte BNT162b1 wesentlich längere Beobachtungszeit (Anwendungsbeginn 23.4.2020) und zwei höhere Dosierungskohorten, kodierte jedoch nur ein Bruchteil des Spikeproteins27. b1 ist 1262 Nukleotiden lang28, b2 428329. Wenn man die höheren Dosierungsgruppen (50 & 60 ug) auslässt, hatte b1 außerdem geringere Nebenwirkungen und vergleichbare Immunogenität, aber wie inzwischen klar ist hat die Entscheidung zugunsten von b2 die Erhebung dieser Daten gar nicht mehr mitgekriegt. Der Zeitraum reicht nur für die Verabreichung einer Dosis; die ersten beiden Geimpften konnten sich erst am 29.6. die zweite Abholen.
In der vermeintlichen “first-in-human”-Studie BNT162-01 unter der Obhut des Paul-Ehrlich-Instituts war die erste Zweitdosis am 6.7.2020 verfügbar, also haben die von BioNTech selbst erhobenen Daten eine noch geringere Rolle gespielt. Dafür gibt es weitere prägnante Beispiele wie etwa der Pfizer-Vorschlag (und FDA-Zustimmung) für Studie BNT162-01 nur einen “gekürzten” klinischen Bericht vorzulegen30, oder die um ein Jahr verzögerte Auswertung wichtiger BNT162-01-Studienproben31 unter dem Vorwand der Auslastung durch Pfizer-Studien.
Das Spektrum der nicht-klinischen Entwicklungsarbeit ist in dieser Hinsicht entlarvend. Von den fünf verfügbaren Studien, die vor der Humandosierung begonnen wurden, beziehen sich drei ausschliesslich auf BNT162b2v9. Zwar wurde 10741 wahrscheinlich wie 10671 mit mehreren Kandidaten durchgeführt, dann aber nur b2-Daten eingereicht - was der ”b1 war nur eine Fassade”-Vermutung nur Nachdruck verleiht.
Aus den Frühjahr-2020-Marketingunterlagen32 von BioNTech lässt sich unschwer herauslesen, dass nicht modRNA, sondern saRNA der eigentlich bevorzugte Kandidat war. Eine Dosis statt zwei, geringere Wirkstoffmenge notwendig, noch längere Proteinproduktion - selbst-amplifizierend halt. Seit April 2023 sind im EU-Studienregister Datensätze zu den 96 c2-Probanden veröffentlicht, und die Ergebnisse sind beeindruckend katastrophal33; bei neutralisierenden Antikörpen hat nur die höchstdosierte Kohorte mit 2x 3ug überhaupt eine messbare Abweichung vom Basiswert. Da diese Kohorte aber auch den einzigen Covid-Fall der ganzen Studie hatte ist es ohne detaillierte Dateneinsicht nicht mal sicher, ob das von der Injektion kommt.
Besonders spannend wird es, wenn die BNT162a1-Daten hinzugezogen werden (ebenfalls bislang nur in dem Studienregister-Link verfügbar). BNT162a1 ist nach zweieinhalb Kohorten (12+12+6 Probanden) wegen Nebenwirkungen zurückgezogen worden34, und hat was neutralisierende Antikörper angeht auch kläglichst versagt. a1 und c2 sind beides uRNAs, Biontech-Bezeichnung für mRNA, die nicht mit N1-Methylpseudouridine (m1Ψ) sondern mit Uracil formuliert werden, und c2 ist zusätzlich eine saRNA, sprich enthält ausser der Sequenz für das Spikeprotein auch angepasste retrovirale Sequenzen, die sich selbst und die Spikesequenz vervielfätigen35. b1 und b2 sind modRNAs, mit dem Nobelpreis-Mechanismus, den zufälligerweise Moderna auch benutzt. b2 benutzt sogar das gleiche Antigen wie Moderna’s mRNA-1273, und seit der Formulierungsänderung von PBS zu Tris sogar das gleiche Buffersystem36. BioNTech’s Eigengewächse erwiesen sich als verstörend grobe Fehleinschätzungen, sollten sie nicht wie b1 nur ein Feigenblatt für b2 gewesen sein. Vielleicht doch nicht “Made in Germany” wie so gerne behauptet, sondern eher “DoD & Fauci”?

Prozess 2
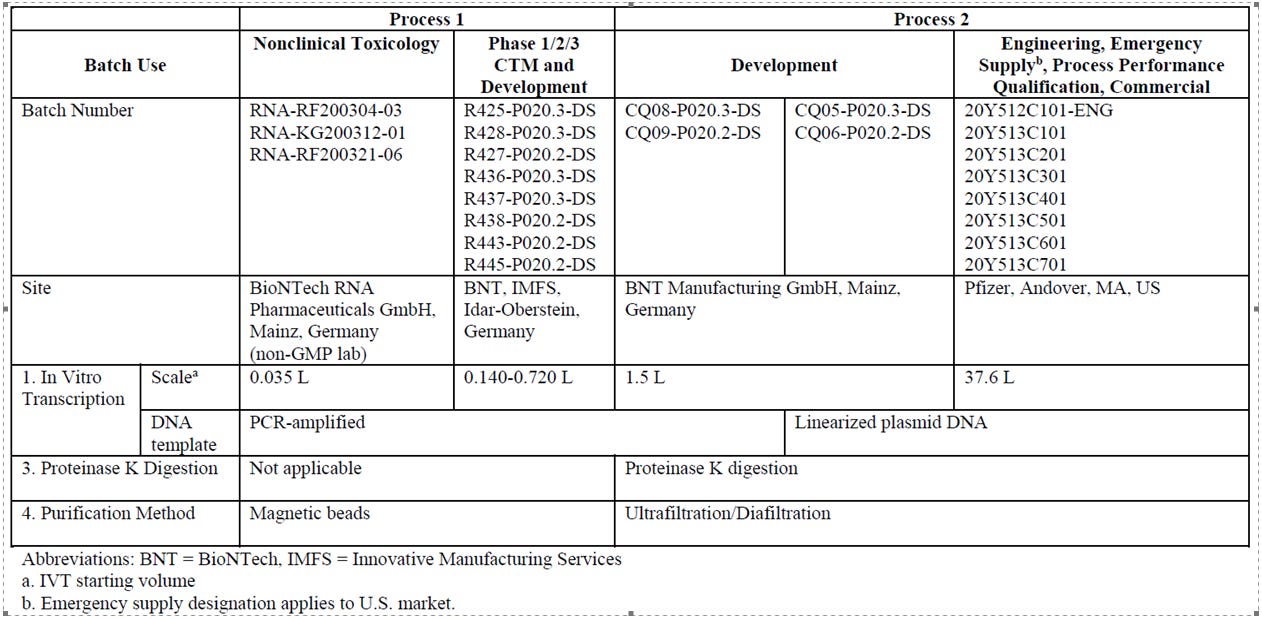
Die Impfstoffe für die beiden klinischen Studien BNT162-01 und C4591001 wurden mit dem “Prozess 1”-Verfahren hergestellt - zell-freie in-vitro Transkription, hohe Integrität der mRNA, hohe Reinheit, winzige Produktionsmenge37.
Pfizer-BioNTech hat im Laufe der kommerziellen Skalierung das Herstellungsverfahren grundlegend verändert, zu Prozess 238 - riesige Bioreaktoren (sprich Fässer) mit mindestens 37L und bis zu ~200L39 genetisch modifizierter Eschericcia coli Bakterien, denen per Hitzeschockverfahren ein Plasmid zugefügt wird, welches die mRNA-Sequenz von BNT162b2 enthält. Unter Zufuhr von Nährstoffen und kontrollierten Temperaturbedingungen vermehren sich die Bakterienkulturen exponentiell und damit auch die in ihnen enthaltenen Plasmide. Anschliessend wird die in den Bakterien enthaltene modRNA durch Lysis herausgeholt, aufbereitet und an andere Standorte zur LNP-Formulierung gebracht.
Soweit die Theorie. In der praktischen Umsetzung stellt sich heraus, dass die Aufbereitung der Plasmid-modRNA nicht besonders effektiv war und das kommerzielle Produkt deswegen die EMA-Richtlinie für dsDNA-Verunreigung um das 18-70-fache überschreitet40 in vielen verschiedenen Chargen41 und dass Pfizer/BioNTech den Zulassungsbehörden gegenüber vorenthalten haben, dass mehrere SV-40 Elemente im Plasmid verwendet werden42. In der angegebenen Sequenz ist diese erkennbar, aber Pfizer hat die graphische Darstellung vorsätzlich modifizert. Die Spikesequenz musste manuell annotiert werden, da kein Programm sie kennt, während die industrieweit automatisch beschrifteten SV-40-Sequenzen manuell aus der Plasmid-Karte entfernt wurden. Diese “Verschleierung” ist inzwischen von der EMA43 und Health Canada44 zugegeben worden.
Bereits am 10.06.45 behauptet Pfizer in einer Einreichung, Impfstoff “auf Risiko” zu produzieren, was in Anbetracht des erst 16 Tage später stattfindenden “Type C”-Treffens doch relativ auffällig ist46. Da die Liste der produzierten DS-Chargen47 zwischen 29.5 und 20.6 eine Lücke aufweist, könnte hier von einer defekten und verworfenen Charge die Rede sein. Im C4591001-Studienprotokoll erscheint Prozess 2 erst Monate später, in Amendment 7 vom 6.10.202048. Um die Vergleichbarkeit zwischen den Herstellungsprozessen zu überprüfen, sollten in einer Unterstudie je 250 Probanden Prozess 2 BNT162b2 oder placebo erhalten. Darüber hinaus sollte nach der EUA-Marktzulassung eine zusätzliche klinische Studie erfolgen. Josh Guetzkow (hier auf substack
) hat wichtige Twitter-Threads zu der Thematik verfasst49. Für alle Details zu den Verunreinigungen ist (Kevin McKernan’s Substack) die zuverlässigste und aktuellste Quelle.Kurz gefasst wurde die klinische Vergleichbarkeit mit je 250 Prozess 2/placebo-Patienten zwar dosiert, seitens der Sponsoren jedoch nie vollständig ausgewertet. Wie eine IFG-Anfrage an die englische Zulassungsbehörde MHRA von N Hunt gestellt und von Josh Guetzkow präsentiert50 ergeben hat, entfernte Protokolländerung 20 im September 2022 dreiundzwanzig Monate später die Verpflichtung, diese Daten auszuwerten, mit der fantastischen Begründung, Prozess 2 sei inzwischen milliardenfach angewendet worden. Das Studienprotokoll ist bislang nur bis Amendment 18 von Sept 2021 verfügbar.51 Der vom Hersteller erzeugte Online-Eintrag im EU-Studienregister, mit der bemerkenswerten Angabe von 81 Todesfällen in der “doppel-geblindeten-aber-nicht-wirklich” Studienphase im Vergleich zu den 38 Stand März 202152, enthält ebenfalls eine Beschreibung auf Protokolländerung 2053 mit identischer Begründung für den Verzicht auf den klinischen Vergleich zwischen den Fertigungsmethoden. Jede der großen Zulassungsbehörden hätte diesem Treiben einen Riegel vorschieben können, jedoch zeigt sich, dass unter deren Aufsicht Pfizer-BioNTech bereits Januar 2021 auf diesen Vergleich verzichtet haben, indem sie eine selbst vorgeschlagene Post-Marketing-Studie54 verändert haben. C4591017 sollte laut ursprünglichem Studienplan vom 18.1.2021 drei US-Prozess 2-Chargen mit einer EU-Prozess 2-Charge und einer fünften “Kontrollcharge” vergleichen. Amendment 1 vom 26.1.202155 entfernt ohne weiteren Kommentar die gesamte Dosierungsgruppe und ersetzt sie mit einer 20ug-Dosierungsgruppe einer der US-Chargen. Die auf der Pfizer-Webseite verfügbaren Zusammenfassungen56 enthalten keinen Verweis auf die entfernte fünfte Dosierungsgruppe, daher ist es noch nicht hundertprozentig sicher, dass es sich bei dieser Gruppe um Prozess 1-Impfstoff handelt. Pfizer-BioNTech haben also die Daten der Probanden, die mit Prozess 2 injiziert wurden, nie ausgewertet, und die Studie, die beide Prozesse vergleichen sollte, sabotiert. Die im Oktober veröffentlichten Unterlagen ermöglichen eine genauere Rekonstruktion der Prozess 2 Vergleichbarkeitsevidenz, die ich besprechen werde, sobald detailliert erarbeitet statt überflogen57.
Am 14.11.2023 wurde auf postvac.org eine äusserst interessante IFG-Antwort der EMA hochgeladen58, zwei Dokumente von Mai 2021 und März 2022 als Antwort auf die Frage, welche “post-marketing”-Auflagen bezüglich DNA-Verunreinigungen bislang erfüllt wurden. Wie auch aus PHMPT-Unterlagen von der FDA hervorgeht, waren kritische Qualitätskontrollen weder validiert noch in manchen Fällen ausreichend beschrieben, und März 2022 lagen noch immer keine Daten vor, welche die Angemessenheit und Effektivität der angewandten DNAse-Enzymaufbereitung belegt. Wie neulich entdeckt Moderna-Patentserien59 eindrucksvoll beschreiben, ist diese DNAse-Aufbereitung alles andere als ideal.
Eine relative neue Entdeckung zu Prozess 260 zeigt wiederholt eine psychopathisch anmutende Verachtung für ethisches Forschungsverhalten. Alle “Prozess 2”-Probanden sind anhand ihrer Randomisierungsnummer identifizierbar, womit die Blindung der gesamten Studie als Lügenkonstrukt entlarvt wird. Es funktioniert folgendermaßen: alle “Prozess 2”-Impfchargen wurden an vier verschiedene Studienstandorte geschickt. An diesen Studienstandorten wurden ab Ankunft der Chargen genau 252 Probanden geimpft, und diese Probanden sind an ihren Standorten die Einzigen, die eine Randomisierungsnummer zwischen 400000 und 499999 haben.

Kritische Datenintegrität
Es ist nicht nur der Fall von Augusto Roux616263, der bei Bekanntwerden direkte strafrechtliche Konsequenzen hätte nach sich ziehen sollen.
Es gab viele Studienstandorte, an denen symptomatische Geimpfte statistisch signifikant seltener getestet wurden64, Standort 1056 z.B. hat genau 0 Tests von Geimpften eingesandt.
Laut einem Schreiben der FDA65 kam man am 25.11.2020 bereits zu dem Schluss, dass die Daten “nicht sauber” sind und die Resultate möglicherweise nicht akkurat, obwohl eine AI-Firma innerhalb von 22 Stunden nach Erreichen der zur primären Effizienzberechnung notwendigen Covid-Fälle die Daten durchgeputzt hat66. Die gleiche AI-Analysefirma hat im Februar 2020 die Zusammenarbeit mit Pfizer begonnen, als Pfizer deren klinisches Studienprogramm LSAC lizensierte67.
Es gibt Hinweise auf manipulierte Phase-1 Antikörper-Daten68, die ein rapides Absinken der Titer verschleiern.
Die erhobenen aber nicht genutzten Nukleokapsid-Antikörper-Laborwerte, die vor Dosierung und einen Monat nach Dosis 2 erhoben wurden, zeigen eine Impfeffektivität von höchstens 53%69, wenn nicht noch wesentlich weniger, wie eine Moderna-Studie vermuten lässt; diese zeigt, dass modRNA-Geimpfte in bis zu 40% der “Impfdurchbrüche” keine Nukleokapsid-Antikörper entwickeln70.
Es ist inzwischen bewiesen, dass Pfizer wesentlich früher von bestimmten Todesfällen wusste, und die Einspeisung in die klinischen Daten absichtlich hinausgezögert hat, womit diese nicht Teil der November-Einreichung wurden71.
Verily, ehemals bekannt als “Google Life Sciences”, war für die Rekrutierung der Studienteilnehmer verantwortlich72, und ICON, die von Pfizer zur Durchführung der C4591001-Studie beauftragte “contract research organisation”, hat bereits April 202073 einen OTA-Vertrag mit dem amerikanischen DoD abgeschlossen. Pfizer hat bis Ende 2020 zwei dieser ”Prototypen-Demonstrations”-Verträge abgeschlossen7475. Augusto Roux hat Dokumentation erstritten, dass der argentinische Arm der Studie unter Militärrecht geführt76 und sogar wegen schwerwiegender Fehler unterbrochen wurde, die Ermittlungen jedoch eingestellt77 und archiviert wurden.
Seit Kurzem verfügbar sind auch die ersten zehn sogenannten SMSRs, die “summary monthly safety reports”, für den Zeitraum von Dezember 2020 bis September 202178. Ausserdem gibt es einen DSUR (development safety update report)79, inzwischen drei PSURs (periodic safety update report)80 und anscheinend gibt es ausserdem einen “bi-monthly” report, von dem jedoch noch kein Exemplar verfügbar ist. Kurz gefasst: Pfizer wurde völlig überflutet, stellte in den ersten sechs Monaten mehr als 2000 zusätzliche Arbeitskräfte ein und brachte es dennoch in keinem dargestellten Monat zustande, die erhaltenen Schadensmeldungen rechtzeitig zu bearbeiten. Bis September 2021 waren 473,000 Geschädigte, 1,7 Millionen Nebenwirkungen und 8383 Todesfälle in der Sicherheitsdatenbank aufgeführt und ein Rückstau von über 70.000 Fällen81. Die einzelnen Berichte sind bis zu 13.000 Seiten lang und zeichnen ein erschütterndes Bild der Interaktion zwischen Zulassungsbehörden und Hersteller; grobe Fehler und billige Verschleierungen in abgebrühter Regelmäßigkeit, Kommentare, Anfragen und Anweisungen seitens der Behörden, die bestürzend verkehrt zur öffentlichen Kommunikation sind, Sicherheitssignale, die eins nach dem anderen dekonstruiert und als unzutreffend geschlossen werden. Hätte die FDA nicht darauf bestanden, wäre das auch mit Myokarditis/Perikarditis geschehen82.

Moderna-Genotoxizitätsstudien
Seit Dezember 2022 ist eine Übersicht über die nichtklinische Studienlage zu Moderna verfügbar83. Die bislang einsehbaren vollständigen Studiendokumente beschreiben unter Anderem Tierversuche aus 2017. Damals war jedoch noch klar, dass es sich um ein GTMP-Impfstoff handelt, dementsprechend sind auch insgesamt sechs Genotoxizitätsstudien enthalten84, vier in vitro und zwei in vivo. In vitro werden die Lipide SM-102 und PEG2000 untersucht, in vivo jeweils ein modRNA-LNP und ein Luziferase-modRNA-LNP-Konstrukt evaluiert. Die einzelnen Studien sind noch nicht verfügbar, lediglich spärliche Bezugnahme auf zusammengefasste Ergebnisse. Die Experimente in Petrischalen waren negativ, die in lebenden Tieren jedoch.. positiv85. Da die systemische Verteilung im Menschen wegen der i.m.-Anwendung begrenzt sei, schätzt Moderna dieses Risiko als gering ein86. Zulassungsbehörden auch, wie an der nach wie vor bestehenden Marktverfügbarkeit des Produktes unschwer zu erkennen.
“The October 2023 batch is supposed to be the second to last batch that is expected to be released for the age 16+ age group, which means there is the possibility that some of the most relevant data will be buried in these final documents.”
Bildbearbeiteter Tabellenausschnitt:

EMA leak Dokument: Annex 1 - Draft 3.2.P.2.2 Drug Product Seite 11, “In order to investigate whether the same mechanism is involved for intramuscular (IM) administration, Luc RNA-containing LNPs comprising ALC-0315 were injected intravenously (0.3 mg/kg) and intramuscularly (0.2 mg/kg) into Apolipoprotein E (ApoE) knockout (KO) mice in the presence (KO+) or absence (KO-) of recombinant human ApoE3.”
EMA Leak Dokument: Rapporteur's Rolling Review Report Quality - COVID-19 mRNA Vaccine BioNTec Seite 56/215
https://www.tga.gov.au/sites/default/files/foi-2389-06.pdf Seite 24 “BNT162b2 (V8) induced lower titre of functional neutralising antibodies (~ 3-5 fold) compared to BNT162b2 (V9).”
https://www.tga.gov.au/sites/default/files/foi-2389-06.pdf Seite 19 “V8 and V9 have identical amino acid sequences of the encoded antigens and differ only in their codon optimisation sequences, which were designed to improve antigen expression (V9) with higher content of cytosine ribonucleotides.”
Siehe Tabelle in Fußnote 10
Siehe Tabelle in Fußnote 10
Experimentbeginn 07.04.2020 in Texas, Charge 09.04.2020 in Österreich bei Polymun hergestellt. Erklärbar dadurch, dass am 7.4. b1-Dosierung begann, die Daten aus dem eingereichten Bericht jedoch entfernt wurden.
Investigator’s Brochure Seite 31
https://phmpt.org/wp-content/uploads/2023/10/125742_S1_M1_meeting-correspondence.pdf Seite 106 “After June 26, 2020 Type C meeting, CBER received clarification information from Pfizer’s Donna Boyce regarding the construct and doses Pfizer is planning to use in Phase 3 (The same information was later submitted to IND 19736, in amendment 24 dated July 1, 2020).”
Biologika-Zulassungsabteilung der FDA “Center for Biologics Evaluation and Research”

meeting-correspondence Seite 497
https://phmpt.org/wp-content/uploads/2023/01/125742_S7_M1_response-08jun2021.pdf “These data were not available at the time of immunogenicity cut-off for this report because the samples were put on hold due to necessary testing prioritizations at the Pfizer labs (eg C4591001 6-month stability and booster; C4591007). Pfizer has now resumed testing of these samples and an updated BNT162-01 study report will be provided once it is available.”
BNT162-01 Studienprotokoll Seite 207, Summary of safety BNT162a1 “Currently, no further dosing with this vaccine candidate is planned”
https://www.tga.gov.au/sites/default/files/foi-2183-09.pdf Seite 13, “The saRNA has two ORFs. The first ORF encodes an alphavirus-derived RNAdependent RNA polymerase (replicase), which upon translation mediates self-amplification of the RNA. The second ORF encodes the vaccine antigen.”
Seite 24, “It is interesting to note that, eliminating the electrolyte buffer and substituting the new organic buffer based on trometamol, the entire formulation of the new PfizerBioNTech preparation called Tris Sucrose becomes, if not identical, at least very similar to that of Moderna's Spikevax vaccine.”
EMA leak Seite 57, Dokument: Rapporteur's Rolling Review Report Quality - COVID-19 mRNA Vaccine BioNTec
Eine Charge in Marburg ergibt bis zu 8 Millionen Dosen (https://investors.biontech.de/news-releases/news-release-details/biontech-provides-update-vaccine-production-status-marburg). Wir wissen, dass 37,5L 1,545,000 Dosen ergibt, was heisst dass die Marburg-Chargen in mindestens 194L-Fässern gezüchtet wurde.
Seite 12, “Ratio of RNA:DNA ranges from 43:1 To 161:1. EMA allowable limit is 3030:1. This is 18-70 fold over the EMA limit.”
Seite 14, “The Sponsor is currently manufacturing vaccine at-risk and is targeting to have US-manufactured and released vaccine doses of approximately (b)(4) available by year-end 2020 with initial deliveries projected for late November.”
Siehe Tabelle in Fußnote 8
Seite 1285, “The initial BNT162b2 was manufactured using “Process 1”; however, “Process 2” was developed to support an increased scale of manufacture. In the study, each lot of “Process 2,” manufactured BNT162b2 will be administered to approximately 250 participants 16 to 55 years of age. The safety and immunogenicity of prophylactic BNT162b2 in individuals 16 to 55 years of age vaccinated with “Process 1” and each lot of “Process 2” study intervention will be described.”

Jeweils Impf-/Placebo:
60/21 (blinded placebo-controlled follow-up Phase 2/3)
38/- Open Label Crossover Follow-up Period (placebo→BNT162b2)
46/- Protocol amendment 18 (boosters)
=165 “Number of deaths (all causes)”

EU CTR C4591001 “15 Sep 2022: Added language pertaining to early completion of this clinical trial as agreed with FDA and EMA. Removed the objective to describe the safety and immunogenicity of prophylactic BNT162b2 in individuals 16 to 55 years of age vaccinated with study intervention produced by manufacturing “Process 1” or “Process 2” because of the volume of BNT162b2 now distributed and administered globally using manufacturing “Process 2,” making this comparison unwarranted.”
meeting-correspondence Seite 40/636, “6.2.3. Clinical Lot Consistency Study: To fulfill CBER’s clinical lot consistency study requirement, a randomized safety and immunogenicity study will compare three lots of the vaccine candidate manufactured with a process suitable for large-scale manufacture and will be performed in 2021.[…] Due to the urgency of the current pandemic and the desire to have a US-licensed vaccine as quickly as possible, the Sponsor proposes that this study be conducted as a post-approval commitment.”
Protocol C4591017 Final Protocol Amendment 2, 03 May 2021
“The study intervention arm “control lot” has been removed”
Assessment Report for the Post-Authorization measure REC 027
Vielen Dank an twitter.com/B_a_f_h_ auf twitter für den Hinweis!
Twitter-Strang zu den Dokumenten
“Moderna's patent application discloses much about the adverse behavior of the plasmid DNA in the body.” Twitter-Strang von PATENT_Sun
“More Moderna Patents that speak to the hazard of residual DNA” Twitter-Strang von Kevin McKernan


Die vielen Ungereimtheiten bei der Pfizer-Zulassungsstudie WELT.de 17.2.2023 Archivlink https://archive.is/m3snI
“The datasets are not clean so results may not be entirely accurate.”
https://icandecide.org/wp-content/uploads/2023/09/28_BLA-125742-0_07-20-2021_Memo_Review-1.pdf
“But thanks to process and technology optimizations, including a new machine learning tool known as Smart Data Query (SDQ), the COVID-19 vaccine clinical trial data was ready to be reviewed a mere 22 hours after meeting the primary efficacy case counts.” https://www.pfizer.com/news/articles/how_a_novel_incubation_sandbox_helped_speed_up_data_analysis_in_pfizer_s_covid_19_vaccine_trial
Saama Technologies, Inc., the number one AI clinical analytics platform company, announced today that it signed an agreement with Pfizer Inc. to develop and deploy an AI-powered analytics solution to reduce the challenges commonly experienced by clinical study data managers and monitors. https://www.businesswire.com/news/home/20200218005688/en/Saama-and-Pfizer-to-Transform-Work-of-Clinical-Data-Managers-and-Monitors-With-AI
Dean Follmann, Holly E. Janes, Olive D. Buhule, et al. Antinucleocapsid Antibodies After SARS-CoV-2 Infection in the Blinded Phase of the Randomized, Placebo-Controlled mRNA-1273 COVID-19 Vaccine Efficacy Clinical Trial. Ann Intern Med.2022;175:1258-1265. [Epub 5 July 2022]. doi:10.7326/M22-1300



https://twitter.com/a_nineties/status/1715785724580995317
Hinweis: der Juli 2021-SMSR ist nur über Download der gesamten Oktober-Produktion verfügbar, der direkte Link auf PHMPT scheint nicht zu funktionieren
SMSR+placebo Exceldatei, Tab SMSR
Seite 3, SMSR April 2021, “Myocarditis and Pericarditis were evaluated and determined not to be risks”
Seite 7438, SMSR Mai 2021, “Closing of the signal myocarditis and pericarditis is not accepted.”
Projekt Lightspeed hat dazu folgendes zu bieten an Zitaten:
"Es gab jedoch einen Nachzügler. Wie B.1 war er auf der modRNA-Plattform aufgebaut, aber statt die kleine RBD des Coronavirus darzustellen, kodierte er für das gesamte Spike-Protein. Dieses Konstrukt hatte Ugur in letzter Minute gegen eine neuere Version, das B2.9, ausgetauscht und die Produktion in Idar-Oberstein umgestellt, nachdem Alex Muiks Tests gezeigt hatten, dass es in Mäusen bessere Antworten hervorrief. Aufgrund der Komplexität des Herstellungsprozesses war es in der ersten Studie am Menschen ganze drei Wochen nach B.1 verabreicht worden. Die Blutproben von Patienten, denen zweimal B2.9 injiziert worden war, würden erst nach einer Weile vorliegen.“ (S. 266)
„Nach jener Videokonferenz, in der Uğur die meisten Entscheidungsbefugnisse abgegeben hatte – was Roshni Bhakta, die Leiterin des Bereichs strategische Partnerschaften und Lizenzen bei BioNTech, damals dazu veranlasst hatte, still in sich hineinzufluchen –, hatte er sich als eines der wenigen Rechte das letzte Wort bei der Auswahl der Kandidaten für die Phase-III-Studie vorbehalten.“ (S. 271)
„Von den zwanzig Kandidaten, die im Februar das Labyrinth des «Projekts Lightspeed» betreten hatten, waren vier klinisch getestet worden. […] Würde das Virus einen Mechanismus entwickeln, um sich dem Zugriff des Immunsystems zu entziehen? B2.9, sagte Ugur damals zu seinen Kollegen, sei «so gut, wie es kaum besser geht», um eine spezialisierte Streitmacht zu mobilisieren: Antikörper und T-Zellen. Aber es bestand immer noch eine Chance, dass SARS-CoV-2 das evolutionäre Wettrüsten gewinnen würde. «Wir kennen die andere Seite der Gleichung nicht», sagte Ugur. «Wir wissen nicht, wie sich der Feind verhalten wird.»“ (S. 272)